Plutôt que passer mon temps à réexpliquer ce que c’est… je vais tenter ma propre explication vulgarisée.
Je sais, je sais. Ce n’est pas lecture plaisante. Mais ce serait vraiment important pour moi si vous preniez le temps de le lire! Bien sûr, c’est à la lecture des articles, de l’effet du SEDh sur mon quotidien, à la longue, que vous comprendrez vraiment ce que c’est… et encore… mais le comment du pourquoi, comme on dit… c’est ici que ça commence et que vous pourrez le comprendre.
Je rappelle que je vulgarise et généralise un brin pour rendre le tout plus digestible… et que, comme pour toute chose, tous les médecins ne sont pas tous au fait des informations les plus récentes; de même, vous pouvez tomber sur des informations qui ne sont pas de la dernière fraîcheur. Mon résumé simplifié se base sur des années de lecture d’articles médicaux, mais surtout sur la nouvelle « bible » des syndromes d’Ehlers-Danlos (que j’explique ici), i.e. la mise à jour des connaissances liées aux SED en 2017, tout autant que de commentaires de personnes atteintes… et ma propre expérience, bien sûr.
Alors voilà, je me lance:
Définition
Les syndromes d’Ehlers-Danlos sont un groupe de maladies génétiques, donc on naît avec. Et en général, on n’est pas les seuls à en souffrir dans la famille. Au moins un parent en a, au minimum, quelques symptômes… Mais pas toujours!
C’est aussi de la famille des maladies orphelines : c’est RARE. Dans le sens qu’il n’y aurait que 1000 à 4000 personnes atteintes au Québec (selon le type de SED, la prévalence varie, pour le syndrome d’Ehlers-Danlos hypermobile, on parle d’environ 1/5000). Dans le sens que je dois épeler le nom de la maladie aux infirmières et autres professionnels de la santé, et que rares sont les médecins qui savent « ce que ça mange en hiver »… Dans le sens aussi que ça prend souvent de nombreuses années avant d’obtenir un diagnostic… et qu’il y a souvent erreur de diagnostic pour commencer. Ce fut mon cas.
C’est une maladie des tissus conjonctifs (peau, ligaments, cartilage, etc. Ces tissus constituent le 2/3 de notre corps), plus précisément, une anomalie du collagène et/ou de la ténascine X (découverte qui date de 2010!).
Types
Il y a 3 types principaux de SED (la classification a changé au fil des ans, pour passer de 11 types, à 6 en 1997, à 13 types en 2017… je vous épargne les types les moins courants):
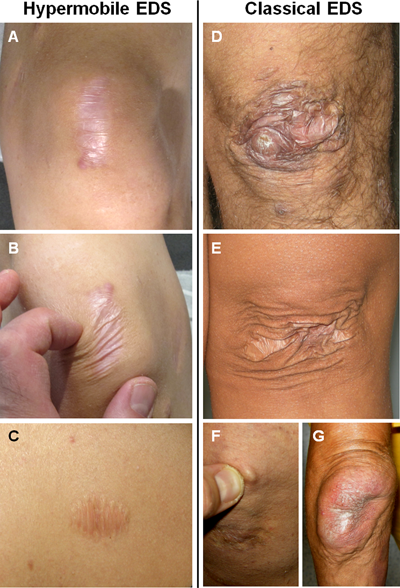
-SED classique (SEDc), qui a surtout des atteintes au niveau de la peau
-SED hypermobile (SEDh) (dont je suis atteinte), où les symptômes les plus importants sont articulaires, reconnu comme le moins grave des trois
-SED vasculaire (SEDv), où il y a des manifestations sévères au niveau artériel, intestinal et utérin, d’où son grand risque de décès.
Symptômes
Les symptômes principaux du SEDh (principaux, parce qu’il y en aurait pour longtemps…), pas nécessairement en ordre:
- Articulations instables (blessures fréquentes et « faciles », par exemple un ligament se déchire partiellement alors que quelqu’un d’autre ne se ferait même pas mal, dislocation en dormant, etc)
- Hypermobilité (duh!)
- Arthrose (ostéoarthrite)
- Douleurs (en général liées aux blessures ou à l’arthrose)
- Fatigue et troubles du sommeil (la douleur empêche de bien dormir et avoir mal est épuisant!)
- Peau fragile et translucide (blessures faciles, cicatrise mal, on voit les veines)
- Syndrome hémorragique (en bon français: on fait des bleus à rien)
- Troubles respiratoires (« faux-asthme », bronchites à répétition)
- Troubles cardio-vasculaires et du système nerveux autonome (pression sanguine irrégulière, pouls irrégulier, difficulté de régulariser sa température, etc)
- Troubles gastro-intestinaux (reflux, constipation ou diarrhée, hernies)
- Migraines / maux de tête
- Troubles dentaires et de l’articulation temporomandibulaire (dislocations, gingivite)
- Troubles cervicaux (colonne vertébrale (instabilité cervicale, dislocations, usure prématurée)
- Troubles gynécologiques (sécheresse de la muqueuse vaginale)
- Dysfonction pelvienne (incontinence, prolapsus des organes pelviens)
- Problèmes psychiatriques (dépression, anxiété)
*Mis à jour selon l’article paru en mars 2017.
…je m’aperçois que je suis atteinte d’au moins trois symptômes/groupes de symptômes de plus qu’au moment de publier la première version de cette page informative, il y a 4 ans, et que mes autres symptômes sont tous plus graves qu’avant. Une belle preuve de la nature dégénérative des syndromes d’Ehlers-Danlos.
Diagnostic
Présentement un SED est diagnostiqué par une entrevue et un tableau clinique. Ça signifie qu’on rencontre un médecin spécialiste qui va analyser le dossier médical, l’historique familiale et peut-être faire faire quelques démonstrations de souplesse…
Si le type classique ou vasculaire sont soupçonnés, une prise de sang sera faite, pour un test d’ADN.
Aucun test n’est fait pour le type hypermobile car aucun marqueur génétique n’a encore été trouvé.
…cependant ces tests sont rarement faits. Parce que ce ne sont pas tous les médecins qui connaissent assez bien la maladie pour poser des diagnostics clairs (le premier médecin qui m’a officiellement diagnostiqué, par exemple, l’a fait du bout des lèvres, croyant que je devais avoir l’air d’une contorsionniste du cirque pour me « qualifier » et utilisait des critères diagnostics dépassés… le diagnostic a été confirmé et re-confirmé par plusieurs médecins depuis… pas que j’en doutais!).
Mais surtout parce que ces tests, qui coûtent très cher… ne sont pas fiables! Pas fiables en ce sens que l’absence de marqueur génétique ne signifie pas absence de diagnostic! Tous les gènes responsables des SED n’ont pas été identifiés, et beaucoup de patients ont eu des tests génétiques négatifs (ils n’ont aucun des marqueurs), mais puisqu’ils ont tous les critères diagnostics, ils ont quand même le diagnostic de syndrome d’Ehlers-Danlos! Selon les données les plus récentes, jusqu’à 10% des personnes atteintes du SED classique n’auraient aucun des marqueurs génétiques associés, par exemple.
Parfois, une biopsie est faite afin de tenter de voir si la peau a des traits de trouble du collagène, typiques dans le SEDc. Mais c’est rarement concluant donc on le fait rarement.
Mon généticien m’a fait tester pour deux des gènes du SEDc, pour la maladie de Stickler et pour la ténascine-X. Nous avons pu éliminer ceux-là. Ça n’élimine pas du tout mon diagnostic de SEDh (mais certains médecins qui n’y comprennent rien me lancent « mais vous ne l’avez pas, ce syndrome, ça dit ici que le test génétique était négatif! »)…
Pronostic et traitement
C’est incurable, dans le sens de « pas de guérison », pas de traitement-miracle.
Avec le type hypermobile, heureusement, le décès est rare… mais la faiblesse veineuse existe et le risque de rupture artérielle est toujours possible, tout comme les complications liées à une maladie associée, surtout que le système immunitaire est affaibli.
Côté traitement, on ne peut donc que soulager les symptômes, et vu les nombreux systèmes touchés par la maladie, les options sont souvent limitées, les manipulations chiropratiques étant carrément dangereuses, par exemple. Les chirurgies sont aussi à éviter et je suis une belle preuve qu’elles sont souvent des échecs (elles demeurent cependant parfois la seule solution).
N’hésitez pas à poser des questions si j’ai oublié quelque chose ou si vous voulez plus de détails sur un point en particulier!
